This was a side project out of curiosity when I was doing my thesis on CHD GWAS in multiethnic populations.
Background
The Human Genome Project (HGP) completed in 2003 has revealed the structure, organization and function of the complete set of human genes. The improvements in high-throughput sequencing technology (“next-gen” sequencing platforms) have sharply reduced the cost of sequencing. The 1000 genome project (1KGP) was initiated in 2007 with the guidance of human gene road map from the HGP and support of the breakthrough in sequencing. The goals of the 1KGP were to discover genetic variants with frequency as low as 1% across human genome and 0.1-0.5% in gene regions, and to characterize the linkage disequilibrium patterns in various ancestral groups. (Durbin 2010). The genomes of 1092 individuals from 14 populations were sequenced in the Phase 1 of the 1KGP (McVean 2012). These sequencing results provided a validated haplotype map of 38 million single nucleotide polymorphisms (SNPs), 1.4 million short insertions and deletions, and more than 14000 large deletions (McVean 2012).
Linkage disequilibrium is an interesting phenomenon in a genomic study. Alleles of SNPs in close proximity on the same chromosome are correlated during reproduction. Random recombination events within a population will break apart chromosome segments, forming ancestry specific linkage disequilibrium patterns unique to the sample population. The systematic difference in allele frequency across populations can result in false positive discoveries in disease studies (Price 2006). Principal component analysis (PCA) is the common method used to correct for the ancestry specific linkage disequilibrium pattern, or population structure in a genome-wide association study (GWAS) (Patterson 2006).
In this report, I performed principal component analysis on the 1KGP data. I am interested in how sensitive the PCA is in terms of distinguishing individuals from various genetic backgrounds.
Methods and Materials
The 1KGP phase 1 genetic data on 1092 individuals were downloaded from the project website (http://www.1000genomes.org/data). VCFtools is used to extract 7714 ancestry informative marker SNPs from the genome-sequencing data, converting zipped VCF files to binary files. PLINK is used to merge the binary genetic data on each SNP into fam/bim/bed files. The first 10 principal components were calculated using SmartPCA, based on the algorithm embedded in EIGENSOFT (Price 2006). The PCA results were merged with the individual list, and plotted in Python.
Results
The table below summarizes the population in the phase 1 of the 1000 genome project by their race and ethnicity.
| Race | Ethnicity | Description | Size |
|---|---|---|---|
| AFR | ASW | Americans of African Ancestry in SW USA | 61 |
| AFR | LWK | Luhya in Webuye, Kenya | 97 |
| AFR | YRI | Yoruba in Ibadan, Nigeria | 88 |
| AMR | CLM | Colombians from Medellin, Colombia | 60 |
| AMR | MXL | Mexican Ancestry from Los Angeles USA | 66 |
| AMR | PUR | Puerto Ricans from Puerto Rico | 55 |
| ASN | CHB | Han Chinese in Bejing, China | 97 |
| ASN | CHS | Southern Han Chinese | 100 |
| ASN | JPT | Japanese in Tokyo, Japan | 89 |
| EUR | CEU | Utah Residents (CEPH) with European Ancestry | 85 |
| EUR | FIN | Finnish in Finland | 93 |
| EUR | GBR | British in England and Scotland | 89 |
| EUR | IBS | Iberian Population in Spain | 14 |
| EUR | TSI | Toscani in Italia | 98 |


The left plot above (Figure 1, left) depicts the first vs. second principal components (PC1 vs. PC2), stratified by race. We can see that the four groups, Caucasians (EUR, blue), Asians (ASN, red), Latinos (AMR, green), and Africans (AFR, black) were well separated. The Asians are especially tightly congregated compared to the other three groups. Latinos spread amid the Asians, Caucasians, and Africans, suggesting a similarity to all three groups. There is a tail of Africans pointing towards the Caucasians, which could be the admixture population between the two groups. The right plot above (Figure 1, right) shows PC9 vs. PC10, stratified by race. The points are evenly scattered on the plot, indicating that there is hardly any segregation by race or systematic ancestry differences. We can also portray the four races on a 3-dimentional scale as shown in the plot below (Figure 2).
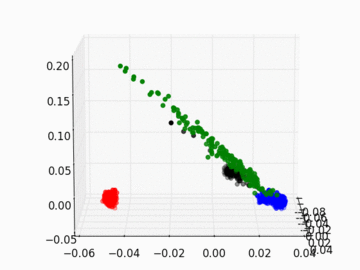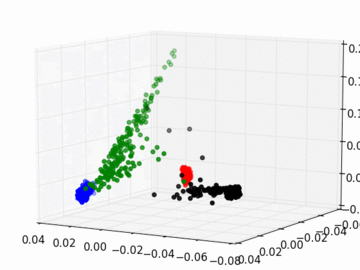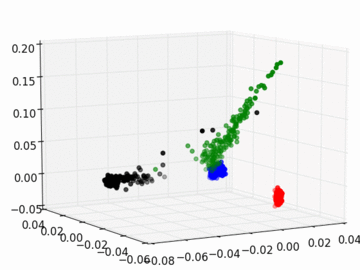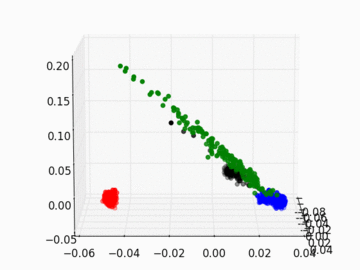
Figure 2 PC1, PC2, and PC3 Plot by Race
Discussion
Majority of the population are Caucasians from Europe, with the exception of Utah residents from the United States. There are too few Spanish to make a conclusion about their genetic LD pattern. Chinese were identified into two groups: Han Chinese in Beijing, and Southern Han Chinese, who reside in the same country, but are fairly distant from each other geographically.
Four race groups are clearly segregated on the left plot of Figure 1. This suggests that principal component analysis is capable of picking up the systematic difference in the genetic structures across racial groups. It is easier to separate Asians, Europeans, and Africans from each other since they are located fairly isolated at the corners of the plot. It may, however, be a challenge to single out Latinos by the values of their principal components. The Latinos carry ancestral inheritance from Asian and European genetic mixture, which results in a diverse genetic background. This is confirmed by the widely scattered AMR PC points on the plot. Figure 2 puts the four groups on a 3D space. We can see that while Latinos carry strong resemblance to Europeans and Asians, they are deviated from Asians on the third dimension, which could be the result of generations of recombination after the mixture.
The right plot of Figure 1 shows that after the adjustment of first 9 principal components, there are no visible differences or segregations on the distribution of PC points by racial groups. This indicates that the first 10 principal components have sufficiently captured the underlying genetic structure of each racial group. The linkage disequilibrium is therefore neutralized when principal components are included in the statistical model.
Conclusions
The principal component analysis is capable of capturing the linkage disequilibrium pattern across populations, and account for the underlying genetic structure in a statistical analysis. It may not be a reliable method to classify individuals by ethnicity due to the interference of admixtures across populations.
References
Durbin, R. M. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
McVean, G. A. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Novembre, J. et al. Genes mirror geography within Europe. Nature 456, 98–101 (2008).
Patterson, N., Price, A. L. & Reich, D. Population Structure and Eigenanalysis. PLoS Genetics 2, e190 (2006).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38, 904–909 (2006).